The Medical Minute: Understanding hypertrophic cardiomyopathy
Although an estimated one in every 500 people is living with an inherited disease known as hypertrophic cardiomyopathy (HCM), many of them may experience few or no symptoms. For some, that could delay diagnoses or bring an awareness of their risk when it’s too late.
“Unfortunately, one of the presenting signs of this disease is sudden death,” said Dr. Eric Popjes, a Penn State Heart and Vascular Institute cardiologist, adding that HCM is the most common cause of sudden death in young athletes in the U.S.. “I have several patients who have family members who died suddenly, which sparked a full evaluation of other people in their families.”
What is hypertrophic cardiomyopathy?
HCM occurs when the heart muscle becomes thickened, reducing its ability to pump blood. Although patients can be diagnosed with the illness at any age, many are teenagers and young adults when symptoms appear.
“More often than not, the most common symptom is feeling short of breath and having exercise limitations,” Popjes said. In addition, patients may experience lightheadedness, fainting, fast heartbeats or chest pain. Some may be found to have a heart murmur on physical exam.
How is hypertrophic cardiomyopathy diagnosed?
Talking with the patient, finding out symptoms and asking about family history is always the first step and frequently the most important part in a diagnosis, Popjes said. Testing with an electrocardiogram or echocardiogram often comes next.
“With a patient who has hypertrophic cardiomyopathy or potentially the genetics behind it, those tests can pick up most cases,” he said. “We also do stress tests and cardiac MRIs that provide very detailed images.”
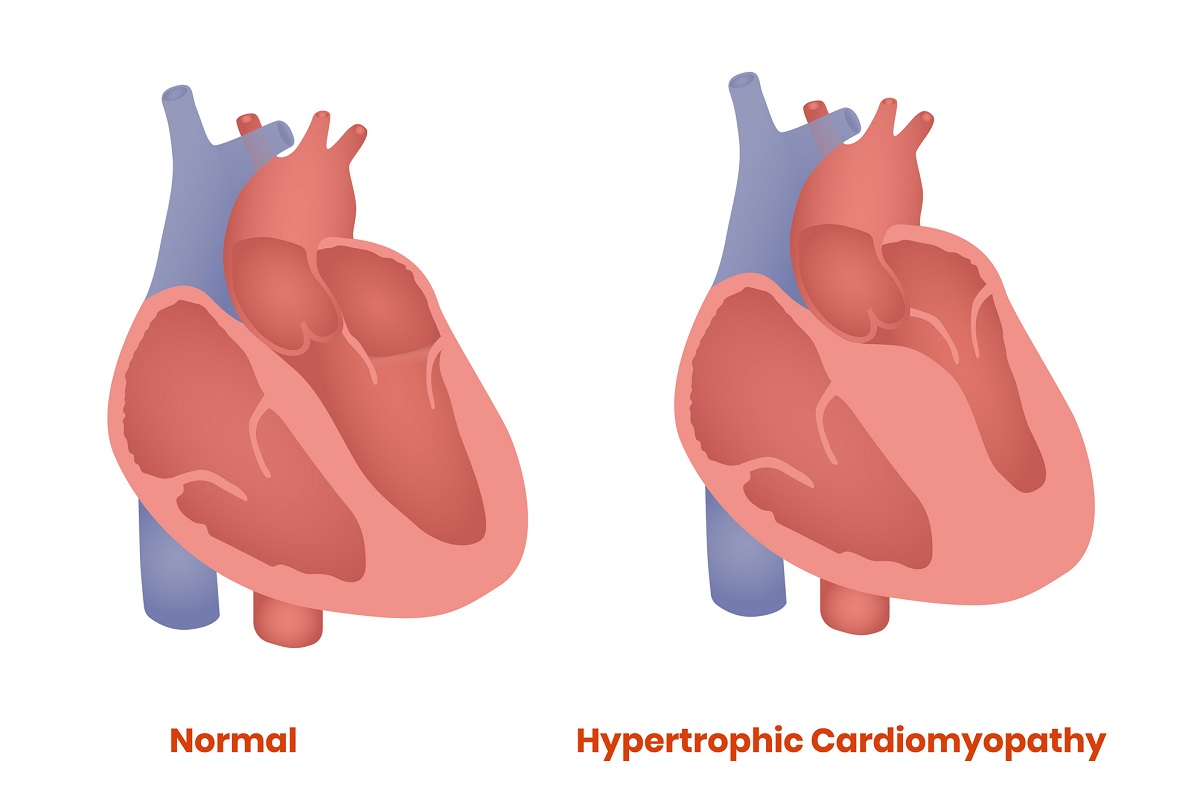
Is hypertrophic cardiomyopathy always genetic?
Although other conditions such as high blood pressure and valve problems can paint a similar clinical picture, HCM is frequently passed down through families.
According to the U.S. Centers for Disease Control and Prevention, a child of an affected parent has a 50 percent chance of inheriting the genetics that can cause HCM. In addition, other family members also have an increased risk making it important for parents, children and siblings of a person with HCM to get screened as well.
“Screening when a family member is diagnosed is very important,” Popjes said. “Genetic testing opens the door to possibly picking up disease in them sooner, allowing us to keep a closer eye on them and preventing bad outcomes.”
How is hypertrophic cardiomyopathy treated?
While many people with undiagnosed HCM may live for years without symptoms or only experience minor symptoms that go unevaluated, they should eventually undergo evaluation and treatment. The good news is that the vast majority of people with HCM are treated with medications.
One of the newer medications is called mavacamten. Approved by the U.S. Food and Drug Administration in 2022, it works by targeting and blocking a protein called myosin, which is essential for muscle contraction. In people with HCM, too much myosin activity causes the heart muscle to contract excessively. Mavacamten reduces this overactivity, helping the heart relax and improving its ability to fill with blood between beats. By improving heart function and reducing symptoms like shortness of breath and fatigue, mavacamten helps improve the quality of life for people with HCM.
“However, it has drawbacks,” Popjes said. “It’s expensive and has significant interactions with other medications that need attention and consideration. Also, patients who take mavacamten need regular echocardiograms to make sure the heart function isn’t being overly suppressed.”
When medications are not enough, patients may benefit from an implantable cardioverter-defibrillator, surgery to remove a portion of the heart muscle or a catheter-based procedure to reduce the degree of heart thickening in order to reduce their symptoms, or even a transplant or insertion of a mechanical heart pump. Helping to guide that care is a team of specialists with expertise treating patients with HCM.
“I need surgeons to weigh in, electrophysiologists to treat rhythm problems, a genetic counselor to discuss test results and a nurse to help coordinate care.” Popjes said. “It is a team effort of expertise on all levels.”
What is the prognosis for HCM patients?
With the appropriate treatment and follow-up care, many patients with HCM can expect to live a normal lifespan with few limitations or complications.
“Someone without symptoms and reasonably good heart function might be able to participate in most physical activities, while others who have any manifestations on their echocardiogram or EKG shouldn’t participate in competitive athletics or really rigorous activity,” Popjes said, adding that modest activity is almost always appropriate. “For patients at high risk we advise them to go out for good walks and live a healthy lifestyle otherwise. They have to be certain they don’t do too much.”
Patients also can benefit from resources such as the Hypertrophic Cardiomyopathy Association, which provides support, advocacy and education.
“The Hypertrophic Cardiomyopathy Association has connections with recognized Centers of Excellence throughout the country that care for patients on a regular basis,” Popjes said. “They’ve done a lot of good work over the years educating patients and getting them referred to care.”
Related content:
- The Medical Minute: Treatment options for heart failure
- The Medical Minute: Aortic dissection starts a race against the clock
The Medical Minute is a weekly health news feature produced by Penn State Health. Articles feature the expertise of faculty, physicians and staff, and are designed to offer timely, relevant health information of interest to a broad audience.
If you're having trouble accessing this content, or would like it in another format, please email Penn State Health Marketing & Communications.